Authors: Bhupendra Singh Punwar1, Chet Ram2, Ankit Singh1 and Ashish Vala13
1 National research centre on plant biotechnology
2 National Bureau of Plant Genetic Resources
3 Anand Agricultural University
In the recent years, development of novel techniques in the life sciences have revolutionized applied research. Genome editing is one of these techniques. The first report on genome manipulation was published by Smithies et al., in 1985 followed by Thomas et al., in 1986. They successfully inserted gene into mammalian genome using homologous recombination. After that lots of reports on genome manipulation were published by various scientific groups on different organisms like baker's yeast, Drosophila etc. But due to non specific or undesirable insertion into the genome, the task of selecting recombinants becomes difficult. This renders homologus recombination inept and allowing narrow room for applied research. Development of targeted genome manipulation also known as genome editing increases the efficiency of the gene editing and eventually expands their uses in both basic as well as applied research.
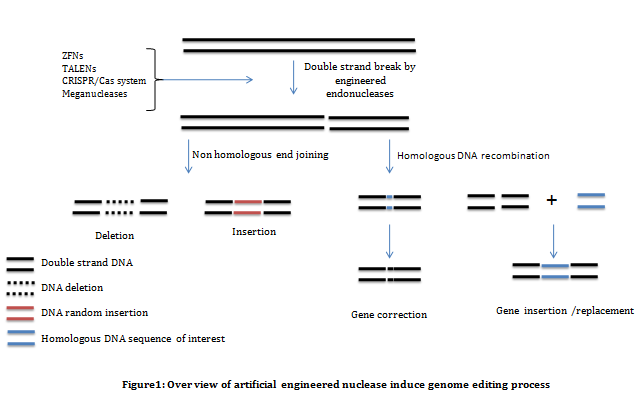
Genome editing is a technique used by genetic engineers to insert, replace, removed or disrupt DNA sequences from the genome using artificially engineered nuclease enzymes or molecular scissors. The concept of genome editing depends upon the understanding of DNA double stranded break (DSB) and its repair mechanisms. DSB repair is one of the most essential mechanisms found in all organisms. Two pathways are found to be in central of DSB repair mechanisms; non-homologous end joining (NHEJ) and homology directed repair (HDR). Non-homologous end joining (NHEJ) directly ligate Double stranded break (DSBs), whereas homologous recombination use sister chromatids as a template for DNA repair. Genome editing completely depends on NHEJ(Figure 1) and hence, for genome editing it is necessary to understand the NHEJ mechanism.
Once DNA strands are broken, double-strand breaks are processed by exonucleases resulting into 3' overhanging single strand tails of about 600 bp. Rad51 and other recombination factors bind to the 3' overhang and locate homologous regions on the sister chromatid resulting in the formation of heteroduplex DNA joints. In NHEJ the broken ends directly rejoin which may cause deletion or insertion of DNA sequences. Therefore, NHEJ ends with disruption of regulatory sequences or gene open reading frame, mostly resulting in gene knock-out.
For NHEJ pathway, the activity of the Ku70/Ku80 heterodimeric protein is essential. The Ku heterodimer binds to the free DNA ends and recruits other protein factors such as DNA-PKcs (DNA dependent protein kinases), XRCC4 to XRCC7 and DNA ligase-IV. In addition to the factors mentioned above, RAD50, MRE11 (Meotic recombination 11 and NBS1 (Nijmegen Breakage syndrome-1) are also involved. As soon as DNA-PK binds to DNA it gets activated and causes the phosphorylation of a number of substrates including p53, Ku, DNA ligase-IV and cofactor XRCC4. Phosphorylation of these factors play a major role in the repair process. Generally before DNA ends ligate, they undergo processing by nucleases and polymerases. The nuclease processing involves the RAD50-MRE11-NBS1 complex, which has exonuclease, endonuclease and helicase activity. The final step of DNA end ligation is performed by a complex of XRCC4, Ku and Ligase-IV.
These are the tools available for genome editing:
a) Zinc finger nucleases (ZFNs)
b) Transcription activator-like effector nucleases (TALENs)
c) CRISPR/Cas system
d) Artificially engineered meganuclease (homing nucleases)
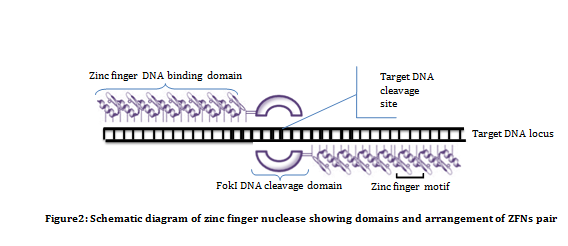
Zinc finger nucleases (ZFNs): ZFNs are artificially engineered enzymes which are a fusion of Zinc finger DNA binding domains to a DNA cleavage domain. Zinc finger DNA binding domain, as name suggests, are involved in DNA binding. The DNA binding domain of ZFNs is a engineered Cys2His2 type arranged in tandem repeats and is designed to bind specific sequences. DNA binding domain of each ZFNs contain three to six individual zinc finger repeats which can recognize sequences between nine to eighteen base pairs. Whereas DNA cleavage domain, involved in restriction of target site, contains domain from the type IIs restriction endonuclease FokI. It requires dimerization to cleave DNA non specifically. Therefore, a pair of ZFNs is required to cleave target sites, and this is achieved by fusing FokI to C-terminal of the ZFN (Figure 2). In order to allow the two cleavage domain to dimerize and cleave DNA, the two ZFNs must bind to opposite strands with some spacer sequence between them. In most of the cases, the length of spacer sequence varies from 5 to 7 bp.
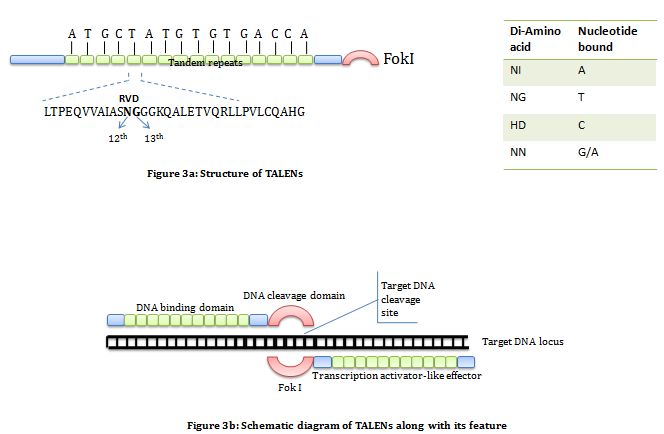
Transcription activator-like effector nucleases (TALENs): It is a artificial enzyme constructed by the fusion of a TAL effector domain with a FokI DNA cleavage domain. The TAL effector domain is a DNA binding domain isolated and characterized from Xanthomonas bacteria. It consists of repeated highly conserved 33-34 amino acid sequence of which the amino acid at position 12th and 13th are variable also known as RVD (Repeat Variable Diresidue). These variable region decide nucleotide specificity. Selecting a combination of repetitive segments containing appropriate RVDs allow us to design sequence specific DNA binding domain. The DNA cleavage domain is non-specific DNA cleavage domain of FokI restriction enzyme. Like ZFNs in TALENs also required pair of nuclease specific for sequence on flanking sites of target (figure 3).
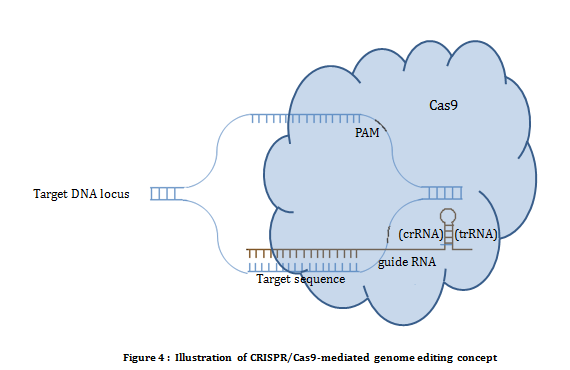
CRISPR/Cas system: The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and Cas (CRISPR associated) system is a prokayote immunity system first discovered in E. Coli. It confers resistance to foreign genetic material such as plasmids and phage. Of the three types of CRISPER mechanisms known, type II is used as a tool in genome editing. Type II CRISPR mechanism requires two components i.e. Cas9 and guide RNA (CRISPR RNA and trans acting crRNA). Cas9 is a endonuclease which introduces double strand break on a site directed by the guide RNA. Endonuclease activity of Cas9 also requires short conserved sequences of 2-5 nt known as protospacer-associated motif (PAM). Using CRISPR/Cas system editing is achived by transfecting/transforming a cell with Cas9 nuclease along with specific guide RNA designed against targeted sequence. Hybridization between guide RNA and genomic sequence in the presence of PAM results into a DNA double strand break (figure 4).
Artificially engineered meganuclease: Meganuclease (Homing endonuclease) are endonucleases with large recognition sites (between 12 to 40bp). Meganucleases are divided into five family based on their sequences and structural motifs viz LAGLIDADG, GIY-YIG, HNH, His-Cys box and PD-(D/E)XK. Among these, the LAGLIDADG family is a valuable tool for genome editing. Using protein engineering approaches, the sequence specificity of meganuclease can be modified to target particular DNA sequences and introduce double-strand breaks.
References:
1. Thomas Gaj, Charles A. Gersbach and Carlos F. Barbas (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering Trends in Biotechnology 31(7) 397-405
2. http://www.technologyreview.com/featuredstory/526511/genome-editing/
About Author / Additional Info:
I am currently pursuing Ph.D in plant biotechnology from Indian Agricultural Research Institute, New Delhi. I like to exploring an area encompassing Biotechnology and molecular biology, the subject of my interest.