Authors: Nabarun Roy1, Utpal Roy2, Mallar Kanti Nath1
1Department of Agricultural Biotechnology, Assam Agricultural University (AAU), Jorhat, Assam, India-785013
2Department of Plant Breeding and Genetics, AAU, Jorhat, Assam, India-785013
What is mixed viral infection (MVI)?
MVIs caused by attack of more than one virus on a host plant. Viral partners interact synergistically and induce severe symptoms more than single infection. Synergistic interactions are produced predominantly by viruses of separate family. Infection of tobacco by Poty Virus Y from family Potyviridae and Poty virus X from family Alphaflexiviridae results in an enhancement of disease symptoms up to 10-fold higher compared with single infection. MVIs have also been found in maize by sugarcane mosaic virus and maize chlorotic mottle virus causing maize lethal necrosis; in cowpea between Cucumber mosaic virus and black-eye Cowpea mosaic virus etc. Sometimes the virus species or strain in MVI is totally unknown to plant pathologists. To manage MVIs, prompt diagnosis is needful that can aid in faultless identification of the causal agents.
Fast Methods to detect MVIs:
Diagnosis of MVIs caused by unknown virus can’t be done by qRTPCR, ELISA, electron microscopy and sap inoculation of indicator species because they don’t give species level diagnosis, until antibodies, primers, or probes for a particular virus are not known. NGS is a metagenomic approach that provides powerful and speedy front-line screen for viruses followed by data mining with homology-dependent (BLAST- Basic Local Alignment Search Tool) and homology-independent (PFOR-progressive filtering of overlapping small RNAs) computational algorithms. This approach does not require prior knowledge of pathogens and can potentially identify both known and new viruses in a disease sample. The most common platforms of NGS used for detection of MVIs are pyrosequencing (454 Life Sciences) or illumina dye sequencing. NGS has been applied to plant virology since 2009 and more than 100 novel DNA and RNA plant viruses from different genera and families have been reported in the recent years by this method.
Approaches of NGS to detect MVIs:
• Enriching of viral RNA by increasing the amount of dsRNA and sequencing of this dsRNA fraction
• Deep sequencing of isolated total nucleic acid from infected host and virus like particles (VLPs)
• Vector enabled metagenomics study for the viruses transmitted through vectors
Bioinformatics tools used in NGS:
Majority of novel viruses discovered using BLAST that compare detected nucleotide sequences (processed contigs) to those in Genbank, and rely on the fact that novel viruses have some homology to known viruses. MEGA (Molecular Evolutionary Genetics Analysis) software, Clustal Omega online tool can be used to assign the reads to possible taxa i.e. it constructs phylogenetic trees. Computer programs such as USEARCH and Hhbits are developed which are faster than BLAST.
NGS data can also be analyzed in Galaxy and PLAN. VirFind, MetaVir and Virome are automated bioinformatics tools specifically used for virus detection and discovery. If there is no sequence homology information known about a new virus/viroid, then homology independent computational algorithm PFOR and PFOR2 can be used. This approach relies on the observations that rolling-circle replication of viroids and some satellite RNAs produces multimeric head-to-tail dsRNAs and that continuous overlapping sets of small interfering RNAs (siRNAs) processed by Dicer from the direct repeat dsRNAs accumulate to high levels in infected plant tissues. PFOR retains viroid-specific siRNAs for genome assembly by progressively eliminating non-overlapping small RNAs and those that overlap but cannot be assembled into a direct repeat RNA. PFOR written in PERL language is extremely slow and is unable to discover those subviral pathogens that do not trigger in vivo accumulation of extensively overlapping small RNAs. PFOR2 written in C++ language is around 8 times faster than PFOR.
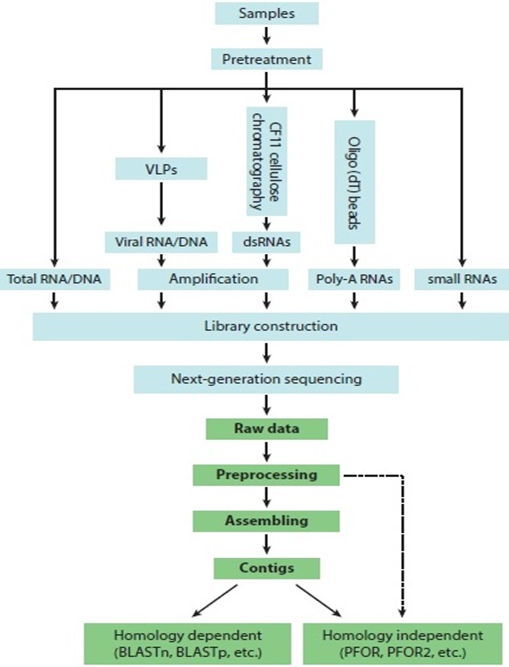
Fig.1: The key steps in the discovery of viruses by next-generation sequencing (NGS)
Conclusion:
Disease outbreak of these mixed infections on certain crops may lead to heavy loss for the farmers. To control these viral diseases, rapid diagnosis is necessary that can help to accurately identify the causal agents. Using of next generation sequencing (NGS) platforms and bioinformatics tools such as BLAST and MEGA can help in the early detection of mixed viral infections. With the help of NGS, sequencing can be done in a non-specific fashion very rapidly, and then sequence similarity and phylogenetic analysis can be checked with the help of already reported sequences using BLAST and MEGA tools of bioinformatics. Thus power of NGS and bioinformatics can be together utilized for the front-line diagnosis of mixed plant diseases that are gaining attention of the researchers globally.
References:
1. Barzon L et al. 2011. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 12: 7861-7884.
2. Prabha K et al. 2013. Applications of next generation high throughput sequencing technologies in characterization, discovery and molecular interaction of plant viruses. Indian J. Virol. 24:157-165.
3. Wu Q et al. 2015. Identification of viruses and viroids by next-generation sequencing and homology- dependent and homology independent algorithms. Annu. Rev. Phytopathol. 53: 425-44.
About Author / Additional Info:
NGS & bioinformatics tools help to detect mixed virus infections in plants