Author: Lalbahadur Singh, Nimmy M. S
Introduction
Plant genomics has a central role in the improvement of crops, including discovery of genetic variation that underlies enhancing performance and increasing the efficiency of plant breeding. Both approaches are important for breeding new varieties and the need to identify new sources of genetic variation. These genomic data are enabling key steps in crop improvement, such as trait identification and alteration, the breeding process and performance optimization, which can now be considered as DNA sequence analysis problems.
Crop plant genomes
The extraordinary diversity of plant species is reflected in their genomes, which vary greatly in size and complexity (Bennett et al., 2011). Dramatic increases in genome size, notably in the grasses, are driven by bursts of DNA-repeat expansion that tend to preserve an underlying conserved order and composition of genes (Bennetzen et al., 2005). DNA repeats have an important role in generating phenotypic diversity and plants have evolved epigenetic mechanisms to limit the parasitic expansion of such repeats (Lisch et al., 2012: Kim et al., 2014). The other dominant feature of plant genome evolution is whole-genome duplication, which is pervasive in most plant lineages (Jiao et al., 2012). Whole-genome duplication can lead to aneuploidy, asymmetric genome evolution (Woodhouse et al., 2010), the rapid loss of genes, exchange between chromosomes and new gene functions, and is therefore an important driver of genetic and phenotypic diversity and adaptation. The large genome sizes, long tracts of related repeat sequences and the closely related homeologous genes in the large gene families of polyploid crops have presented considerable challenges for sequencing technologies. Assembling accurate and representative genomes and assessing the full range of available genetic variation are therefore central aims for crop plant genomics.
DNA sequencing and assembly technologies
A variety of sequencing methods are now available for different applications in crop improvement (green pyramid). The number of genomes that can be sequenced cost-effectively varies according to the method applied (left in figure 1). Long-read technologies from PacBio, alone or coupled with Illumina assemblies, can be used to provide accurate long-range assemblies for a smaller number of genomes. These are used to define comprehensively the range and types of variation that are found in the genomes of a species (the pan-genome). Linked reads, coupled to Illumina sequencing, may provide more cost-effective capacity for sequencing on the order of thousands of genomes, which is useful, for example, for the identification of structural variation. Skim sequencing consists of low-coverage (for example, 5â€"10x) Illumina reads and presents a cost-effective way of identifying genetic variation and haplotypes in populations. Exome sequencing captures gene-coding regions, and genotyping by sequencing typically involves the sequencing of about 100â€"150 bases from a randomly located restriction-enzyme cleavage site in the genome.
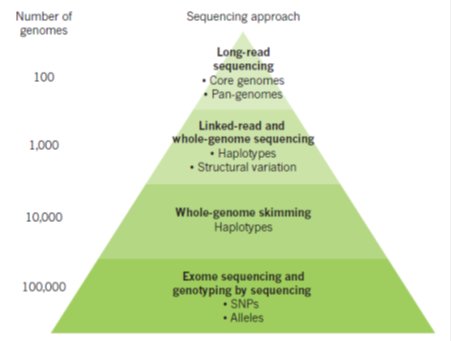
Figure 1: Optimal sequencing systems for crop applications (Source: Bevan et al, 2017)
Erosion of genetic diversity in cultivated crops and its re-incorporation through genomic
Genetic diversity (coloured circles figure 2a) in populations of wild precursors of crops has been eroded by domestication, in which a limited range of diversity is present in landraces that were initially selected and adopted for cultivation. Subsequent breeding has drawn on a limited range of the variation present in landraces to produce the elite cultivars that are used in modern agriculture. The identification of genes for crop improvement can use mutagenesis to introduce changes into the DNA of crops. Mutants with desired characteristics can be identified by screening for desired properties, known as phenotypes. In practice, this method is time-consuming and imprecise unless a specific phenotype can be measured in large populations. Mutant lines with desired phenotypes are pooled and sequenced. Genomics can accelerate the process of identifying mutants by sequencing populations of mutant crops (or a range of wild relatives). Sequencing can be targeted to all genes, or specific families of genes, using sequence capture methods. RNA can also be sequenced to identify changes in gene expression that are caused by mutagenesis. Sequences of mutant lines are then compared to identify genes that are consistently mutated in the lines that exhibit the desired phenotype (figure 2b). Genomics can also be used to access genetic variation in populations of crop wild relatives. A population can be sequenced using a variety of approaches (described in Figure 1). At the same time, the population is screened for a range of phenotypes of interest. Patterns of sequence variation, or haplotypes, can be associated with phenotypes to identify sequence variation that may cause the phenotype (figure 2c).
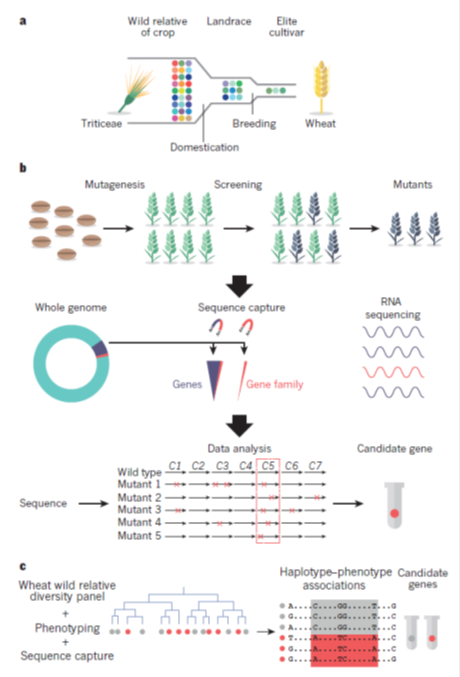
Figure 2: Exploiting genomics to recapture genetic diversity (Source: Bevan et al, 2017)
Concluding remark
Progress in genomics technologies is now enabling the rapid and cost-effective sequencing and assembly of the largest and most complex plant genomes. Researchers can now access and characterize a vast reservoir of natural genetic variation from wild or undomesticated relatives of crops. The application of improved short-read sequencing and genome assembly will continue to provide the most cost-effective solutions for the accurate de novo assembly of larger plant genomes and accessing genetic diversity. However, it is clear that sequencing technologies that involve longer reads, including single-molecule, real-time sequencing, and linked-read sequencing on long molecules will have a major impact by improving sequence assembly and perhaps even by supplanting the short-read sequencing of crop genomes if increases in accuracy and cost-effectiveness can be maintained.
References:
1. Bevan, M. W., Uauy, C., Wulff, B. B. H., Zhou J., Krasileva K., Clark, M. D. (2017). Genomic innovation for crop improvement. Nature, 543, doi:10.1038/nature22011.
2. Bennett, M. D., Leitch, I. J. (2011). Nuclear DNA amounts in angiosperms: targets, trends and tomorrow. Ann. Bot., 107, 467â€"590.
3. Bennetzen, J. L., Ma, J., Devos, K. M. (2005). Mechanisms of recent genome size variation in flowering plants. Ann. Bot., 95, 127â€"132.
4. Lisch, D. (2012). How important are transposons for plant evolution? Nature Rev. Genet., 14, 49â€"61,
5. Kim, M. Y., Zilberman, D. (2014). DNA methylation as a system of plant genomic immunity. Trends Plant Sci., 19, 320â€"326.
6. Jiao, Y. et al. (2012). Ancestral polyploidy in seed plants and angiosperms. Nature, 473, 97â€"100.
7. Woodhouse, M. R. et al. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol., 8, e1000409.
About Author / Additional Info:
1, Right now I am pursuing Ph.D. in biotechnology from Indian agricultural research institute (IARI), new delhi. Currently I am working in the area of miRNAs in pulse crop.
2, Scientist, NRCPB, IARI, new delhi