Author: Ila Ruchie
GASTRIC CARCINOGENESIS
Gastric carcinogenesis progresses stepwise from normal mucosa/superficial gastritis, atrophic gastritis (GA) to gastric cancer. Gastric cancer ranks fifth in GLOBOCON 2012(cancer fact sheets) among the most common cancers in prevalence, and is caused by Heliocobacter pylori its main etiological factor. Helicobacter pylori, a Gram-negative bacterial species selectively colonizes gastric epithelium. Bacterial, environmental and host genetic factors combine to define the degree of gastric damage. Interestingly, H. pylori infection is considered a necessary but insufficient cause for gastric carcinogenesis, as the presence of the infection does not automatically result in a high incidence of gastric cancer. Helicobacter pylori colonizes the majority of persons worldwide, and the ensuing gastric inflammatory response is the strongest singular risk factor from peptic ulceration to gastric cancer. However, only a fraction of colonized individuals ever develop clinically significant outcomes like gastric cancer. Disease risk is combinatorial and can be modified by bacterial factors, host responses, and/or specific interactions between host and microbe. Gastric carcinogenesis progresses stepwise from normal stomach, inflammation and precancerous conditions to carcinoma, as described by Correa’s cascade (Correa P. (1992). In this cascade, host genetic and environmental factors interact to modulate the transition from one step to the next. Therefore, to understand the carcinogenesis process, it is necessary to identify the host genetic components that play crucial roles in each transitional step, namely, normal mucosa/superficial gastritis (NOR/GS)->atrophic gastritis (GA)->carcinoma transition.
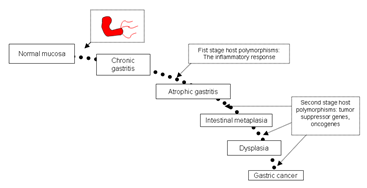
Figure 1. Modified Correa’s cascade (till date): The progression of gastric carcinoma and significance of H.pylori and host genetic factors in the process (Garza-Gonzalez and Perez-Perez, 2013).
RELEVANCE OF HOST GENETIC FACTORS IN GASTRIC CARCINOGENESIS
Peek et al. in 2002 via nature wrote that there has been evidence indicating H. pylori is capable of sending and receiving signals from cellular components within the gastric mucosa, allowing host and bacteria to participate in a dynamic equilibrium. Sustained interactions between H. pylori and humans significantly increase the risk for atrophic gastritis, intestinal metaplasia, and distal gastric adenocarcinoma. Several recent studies proposed a distinct sequence for H. pylori exerting its virulence in the host stomach: (i) adhering to and colonizing the surface of gastric epithelial cells, (ii) evading and attenuating the host defense, and (iii) invading and damaging the gastric mucosa. Several key issues that still need to be clarified, such as which virulence factors of H. pylori are involved in the three pathogenic steps, which host genes respond to the step-specific virulence factors, and whether and/or how the corresponding host genetic variations influence the risk of gastric carcinogenesis.
According to El-Omar et al., 2008, H. pylori bacterial virulence factors as well as host genetic factors play a major role in orchestrating the increased risk of cancer. The study of such hostâ€"bacterial interaction is key to uncover the molecular and cellular pathways involved and will ultimately lead to developing preventive and therapeutic strategies against this global killer. The differences in prevalence of gastric cancer have been explained as a multifactorial process with an interaction involving both infection with Helicobacter pylori as a triggering factor and host genetic susceptibility as an important explanation for inter individual variation in gastric cancer risk. Host factors in particular, genetic polymorphism in adaptive and innate immunity response gene sseem to increase the risk of gastric cancer largely through the induction of severe gastritis.
Host genetic polymorphism study may help identify individuals at high risk of developing gastric cancer so that early intervention may be taken to help prevent its development. Recently, El Omar et al. presented genetic and biologic evidence highlighted the important roles of H.pylori-related genetic variations in modulating the H.pylori pathogenicity, thus influencing the host’s susceptibility to cancer. A classic example of this paradigm is the study of a Caucasian population. El-Omar et al. in 2000 identified two promoter polymorphisms (rs16944 and rs1143627) in the IL-1B gene that were in strong linkage disequilibrium and were shown to be associated with an increased risk of both hypochlorhydria induced by H.pylori and gastric carcinogenesis.
H. pylori infection induces both interleukin-1Beta (IL-1Beta) and tumour necrosis factor-a (TNF-a) production, and these cytokines inhibit gastric acid secretion, leading to the development of gastric precancerous lesions and cancer (El-Omar et al., 2000). Polymorphisms within the IL1B gene increase IL-1 Beta expression and IL-1ra binds to the IL-1 receptors, modulating the pro-inflammatory effects of IL-1 Beta. Regarding the IL1RN gene, a variable number tandem repeat (VNTR) polymorphism has been detected within intron 2, and five allelic variants have been identified in the number of repeats varying from 2 to 6 (El-Omar, 2001). The ability of H. pylori to infect and remain in the human stomach induces a chronic inflammatory response, which may be of variable magnitude depending on the genetic make-up of the host. Most of the single nucleotide polymorphisms (SNPs) studied are situated in the gene promoter region and play important roles in modulating gene expression.
Caiyun et al., 2014 reviewed that Urease, BabA and SabA are crucial in the adhesion-step, PGN and LPS in the immune evasion-step, and CagA, VacA and TipAlpha in the mucosal damage-step were documented to play an important role in step-specific pathogenicity of H. pylori infection. They also mentioned evidence further supporting a role of potentially functional polymorphisms of host genes directly responding to these pathogenic step-specific virulence factors in the susceptibility of gastric carcinogenesis, especially for urease-interacting HLA class II genes, BabA-interacting MUC1, PGN-interacting NOD1, LPS-interacting TLR4, and CagA-interacting PTPN11 and CDH1. In short this H.pylori bacteria influences and induce conditions of polymorphism which shifts the loyality of our host genetic elements against us, leading to its persistence in our tissue and finally causing gastric cancer.
Future prospective and Conclusion
The polymorphisms in host genetic elements have brought us new prognostic and diagnostic prospective in gastric carcinogenesis research and study. Genome wide association study (GWAS) now a day’s helps in defining a gastric cancer specific genetic risk profile. Genetic profiling combined with H.pylori testing to deliver eradication therapy to those who are at risk and to avoid giving unnecessary antibiotics therapy to those who are not at risk of developing gastric cancer. Such a strategy is the only way to be ahead of this global killer and to abash it down.
Abbreviations
• BabA, blood-group antigen binding adhesion A;
• CagA, cytotoxin-associated antigen A;
• cagPAI, cag pathogenicity island;
• CD74, HLA II-associated invariant chain;
• CLR, C-type lectin receptor;
• IL, interleukin;
• FUT4, fucosyltransferase 4;
• HLA II, human leukocyte antigen class II molecule;
• H. pylori, Helicobacter pylori;
• iE-DAP, Y-D-glutamyl-meso-diaminopimelic acid;
• Leb, Lewis b;
• Lex, Lewis x;
• LPS,lipopolysaccharide;
• LRR, leucine-rich repeats;
• MUC, mucin;
• NCL, nucleolin;
• NLR,NOD-like receptors;
• NOD1, nucleotide binding oligomerization domain containing protein 1;
• PAMP, pathogen-associated molecular pattern;
• PGN, peptidoglycan;
• PRR,pattern recognition receptors;
• PTPN11, protein-tyrosine phosphatase, nonreceptor-type 11;
• RLR, RIG-like receptors;
• RPTP, receptor-like protein tyrosine phosphatase;
• SabA, sialic acid-binding adhesion A;
• Tip Alpha, TNF-a inducing protein;
• T4SS, type IV secretion system;
• TNF-Alpha, inducing protein;
• TLR, Toll-like receptor;
• VacA, vacuolating cytotoxin;
• VNTR, variable number tandem repeat
References
1. Caiyun et al., (2014) Review: Host genetic factors respond to pathogenic step-specific virulence factors of Helicobacter pylori in gastric carcinogenesis. Mutation Research/Reviews in Mutation Research Volume 759, Pages 14â€"26.
2. Correa P. (1992) Human gastric carcinogenesis: a multistep and multifactorial processâ€"First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res., 52, 6735â€"6740.
3. El -Omar et al., (2000). Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature., 404(6776), 398-402.]
4. El-Omar EM. (2001) The importance of interleukin 1beta in Helicobacter pylori associated disease. 48(6):743-7.
5. El-Omar et al., 2008 Helicobacter pylori infection and the pathogenesis of gastric cancer: A paradigm for hostâ€"bacterial interactions. Volume 40, Issue 7, Pages 504â€"509
6. Garza-Gonzalez and Perez-Perez, 2013 Relevance of Host Factors in Gastric Cancer Associated with Helicobacter Pylori.
7. Peek et al.,(2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Rev Cancer 2: 28â€"37.
About Author / Additional Info:
I am a biotechnologist just trying to trace my path in this vast field. I am a learner and believe that learning is a never ending process. There is so much to read, explore and to unravel in biotechnology and in my opinion sharing of this knowledge will help us in doing so.