Whether our blood cells are protector or destroyer? If they are protector, by constant maintenance and immune protectant of all cell type in body, what makes one third of people in world's population to die with myeloid leukemia. This article provides a glimpse about role of BCR-ABL1 oncogene in myeloid of leukemia.
In leukemia, the hematopoietic stem cell undergoes either immature or partial maturation. These cells can reproduce quickly and invade through blood stream which spread to other organ resulting in interruption of their normal function. BCR-ABL1 oncogene is responsible for most cases of chronic myelogenous leukemia and acute lymphoblastic leukemia. BCR-ABL oncogene is the result of t(9;22)(q34;q11) reciprocal translocation generating Philadelphia chromosome.
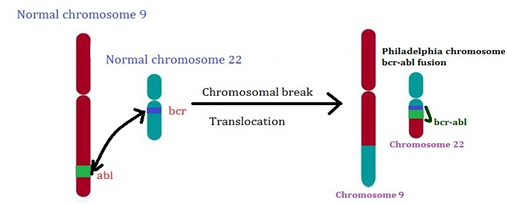
Figure1: Philadelphia chromosome
BCR-ABL gene is located on chromosome 22 and depending upon the fusion, the gene encodes a protein of size between 190 to 230 kDa. Protein has three isoform, wherein 190 kDda protein is being expressed in a condition called acute lymphoblastic leukemia (ALL), 210 kDa protein is related to a condition called chronic myeloid leukemia (CML), and 230 kDa is associated to a condition called chronic neutrophilic leukemia (CNL). The fusion protein helps in leukemic phenotype determinations by regulating lineage commitment.
BCR & ABL1 genes and BCR-ABL1 kinases:
BCR consists of 23 exons in which exon 1 and 2 are alternative exons within first intron, and 3 main breakpoint cluster region m-bcr, M-bcr, and µ-bcr. ABL1 consist of 2 alternative exons, tyrosine kinase domain proceeding with sr c- homology domain.
BCR-ABL1 tyrosine Kinase: The combination of breakpoints within BCR and ABL1 genes generate different fusion transcript encoding protein. SRC and ABL1 kinases share a common central core of overall 42% homology composed of a tyrosine kinase domain, a SRC-homology-2 (SH2) domain, and an SH3 domain. There is significant difference between SRC and ABL1 kinases in upstream and downstream of SH3 and kinase domain. The NH2 terminus in ABL1 and BCR-ABL1 kinases is the "Cap" region. Two isoforms of ABL1 (1a and 1b) are generated by alternative splicing of the first ABL1 exon. ABL1b contains a myristate site (Myr-NH) at the extreme end of the amino-terminal segment, binds to the kinase domain and keeps the SH2-SH3 autoinhibitory structure. The homology region in SRC family kinase is the N-terminal membrane-localization domain which is also referred to as the SH4 domain.
Molecular signaling in BCR-ABL1:
BCR-ABL1 mediated leukemeogenesis is mediated by phosphorylation of BCR Tyr177 which provide docking site for GRB2(growth factor receptor binding receptor-bound protein 2) and binds to GAB2(GRB2 associated binding protein) which activates PI3K and extracellular signalling kinase and SOS resulting in RAS/MAPK activation which in turn activate BCL-2 gene activation(antiapoptotic protein). Phosphorylation of BCR-ABL1 activate PI3K/AKT and interferon consensus sequence binding protein (ICSBP) result in decreased DNA binding resulting in decreased repression of BCL expression and MYC gene which finally result in activation of BCL-X gene transcription which promote cell proliferation and survival of leukemia cell.
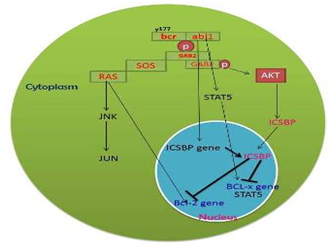
Figure 2: Molecular signaling event in BCR-ABL1 mediated leukogenesis.
Transformation of CP to BP CML:
Chronic myloproliferative phase (CP) to blast phase (BP) result due to progression in disease condition. This is mainly due to BCR-ABL1 expression which activates several transcription factors that regulate growth factor dependence, clonogenicity, protection against apoptosis and motility. Activation of differentiation gene resulting in other genetic aberration, alters the renewal division which promote immature leukemia progenitor cell to BP CML. Production of ROS by BCR-ABL1 results in genomic instability, DNA damage, chromosomal abnormalities and inactivation of tumor suppression genes.
CP CML cell has HSC disorder since BCR-ABL1 gene fails to transform cells lacking inherent self-renewal potential. BCR-ABL activates the expression of the mammalian stress response gene SIRT1 in hematopoietic progenitor cells and that this involves STAT5 signaling. SIRT1 activation promotes CML cell survival and proliferation associated with deacetylation of multiple SIRT1 substrates, including FOXO1, p53, and Ku70 Preleukemic genetic abnormalities by BCR-ABL1 expression, BCL-2 overexpression and loss of JUNB and ICSBP expression help HSC to acquire survival, proliferation and loss in their ability to undergo apoptosis. Further genetic/epigenetic factors provide self-renewal potential by aberrant catenin activation and arrest myeloid differentiation thus CP CML undergo transformation to BP CML.
In short, the ABR gene is associated with tyrosine kinases production, which controls major functions of the cell. Soon after the augmentation with BCR gene, it transcribe a mutated protein kinase. One of major functions of tyrosine kinases is cell cycle control and they are controlled by several co-factors. Mutated tyrosine kinases is continuously in "ON position". They don't require any co-factor activation, resulting in uncontrolled cell division, inhibited DNA repair mechanism and that leads to genomic instability causing leukemia. There are many things which are largely being unanswered. For example, what makes or controls the expression of BCR-ABL1 during prognosis of the disease. Is that disease condition is solely because BCR-ABL1, do any other factors augment the effect of genomic instability, self-renewal, impaired DNA repair mechanism etc. Do they exhibit any positive feedback mechanism for their expression? Likewise many questions comes into the minds of cancer biologists. For the better treatment strategies, we do need more explorations at molecular level of the disease.
References:
1. Salesse S et al., BCR/ABL-mediated increased expression of multiple known and novel genes that may contribute to the pathogenesis of chronic myelogenous leukemia. Mol Cancer Ther. 2003 Feb;2(2):173-82
2. Alfonso Quintas-Cardama et al., Molecular biology of bcr-abl1- positive chronic myeloid leukemia. Blood. 2009 113: 1619-1630
3. Wolfgangwarsch et al., JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia Blood 2013 122:2167-2175.
About Author / Additional Info: