Author: Susmita Das
INTRODUCTION
Cancer is regarded to be one of the deadliest groups of disorders of the decade in all over the world which is characterized by uncontrolled division of cells and rapid growth of them within some parts or a particular site of body. The newly formed cells are known as neoplastic cells with proliferation forming a mass of undifferentiated cells with special properties of neoplasm, metastasis and lack of contact inhibition. In order to classify they are divided according to the tissue they originate from and again can be sub-divided into more than 100 types.
STATISTICS AND PREVALENCE OF CANCER WORLDWIDE AND IN INDIA
According to Globocan reports there was 14.1 million estimated cancer cases worldwide comprising 13% cases of lung cancer, 1.7 millions of breast cancer and 1.4 million of colorectal cancer cases in 2012 [1]. According to the statistics of American Cancer Society in 2016, about 1,685,210 cases of cancers were expected to be diagnosed, with 595,690 Americans were to die of cancer. Proceeded only by heart disease it is the second most common disease in US, with 1 death in every 4 individuals. Additionally more than 60% of new cancer cases are expected to occur in Africa, Asia and Central and South America with 70% deaths among the cases. In India, according to ICMR projection, total number of new cancer cases in 2016 is expected to be around 14.5 lakh comprising of 1.5 lakh cases of breast cancer, 1.4 lakh cases of lung cancer and 1 lakh cases of cervical cancer with a significant increase in rectum and colon cancer due to lifestyle modifications and oral cancer due to betel nut chewing [2].
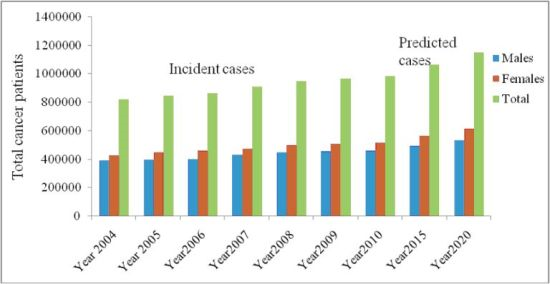
Fig 1: Prevalence of cancer in India (image source: http://www.moneycontrol.com/gestepahead/curing/article/india-and-the-fight-against-cancer-965373-4.html )
AN OVERVIEW OF THE CONVENTIONAL CHEMOTHERAPY
The excessively active growth signaling pathways make them susceptible to wide range of drugs to control over the disseminated disease. Various targets are exemplified as growth signaling molecules, cellular replication and expression and various enzymatic targets. They are mainly classified as below:
- Alkylating agents: Introduce alkyl group into nucleotide creating cross linkage.
- Antimetabolites: Broadly classified as Folate antagonists (inhibition of dihydro folate reductase enzyme (DHFR) interfering with metabolism), Pyrimidine antagonists (Blockage of pyrimidine formation by incorporation in new DNA) and Purine antagonists (Inhibition of S-phase of cell cycle mimicking the activity of purine)
- Intercalating agents: Formation of intercalation between specific base pairs in DNA blocking replication.
- Enzyme inhibitors: Inhibition of topoisomerases (I and II) and resealing of DNA inducing apoptosis.
- Mitotic Inhibitors: Arresting cell division in metaphase by binding with tubulin and stabilization of polymerisation reactions
- Hormonal supplements and analogues: Blocking of signal transduction of normal hormones and feedback inhibition.
- Anti-angiogenesis: Reduction in blood vessel formation in local tissues.
- Antisense therapy: Specific blockage to the gene responsible for cancer formation outlined by gene mapping and backbone modification of nucleotides.
- Apoptosis promoters: Stimulation of apoptosis selectively in tumor cells.
- Cell cycle inhibitors: Inhibition of key steps in cell cycles
- Differentiation stimulator: Differentiation of cells associated with tumors.
- Gene therapy: Replacement of tumor suppressor proteins (p53, pRB etc.)
- Fernesyl transferase inhibitors: Inhibition of attachment of RAS protein.
- Glycosylation inhibitors: Inhibition of metastasis by alteration of sugar pattern on cell surface.
- Monoclonal antibodies: Inhibitor of oncogene encoded receptors or adhesion molecules on surface of tumor cells.
- Protein kinase inhibitors: Inhibition of signal transduction associated with protein kinase.
- Telomerase inhibitors: Inhibition of active Telomeres.
- Vaccines: Boost up T-cell mediated immunity.
RESISTANCE OF CANCER CELLS: A NEW CHALLENGE
Resistance and relapse of cancer impart to be a major impediment in the field of clinical oncology. Being refractory to treatment for cells, lower responsiveness to the treatment and subsidence of the effects of chemotherapy are major concerns depicting the resistance to conventional drug and therapy. Single drug therapy often proves to be failure in such cases and relapse of the symptoms occurs within a few days. In some cases, initially the treatment proves to be beneficial but later the effectiveness subsides. A correlation has been found with the tumor volume and the resistant cells. In a typical case, 1 in 106 - 109 cells can bear resistivity. In case of a tumor, it usually contains 109 cells, so it bears 1-10,000 resistant cells depending on its volume and size. However extensive chemotherapy has some effects on the reduction of mass, but complete elimination is a challenge till date [3].
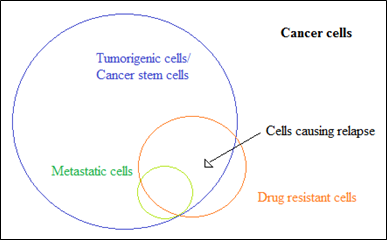
Fig 2: Proportion of resistant cells within complete cancer cells
MECHANISMS OF CANCER RESISTANCES
The mechanisms of chemotherapeutic drug resistances have been well studied till date that highlights influences of some major pathways leading to failure in obtaining the objectives of such therapies. It may be inherent in a subpopulation of a heterogeneous cancer cells and otherwise gradual unresponsiveness develops following the therapy. Although many types of cancers remain initially susceptible to therapy, over time they can develop resistance through drug efflux and DNA mutation and metabolic changes promoting drug inhibition and degradation. Both primary and acquired resistance can be caused by the mechanisms mentioned underneath [3-5].
- Altered membrane transport involving p-glycoprotein and MDR-associated proteins
- Altered target proteins and enzymatic pathway
- Reduction in activation of drug
- Altered expression of enzymes and enhancement of drug degradation in vivo
- Drug inactivation after conjugation with cell metabolites
- Drug interaction and cross resistance
- Enhanced DNA repair and nucleotide excision repair
- Apoptosis defects due to molecular interaction.
MDR is the most discussed and potential obstacle to keep administered drug level below the minimum therapeutic level leading to failure of chemotherapy. Two classes of membrane transporter proteins involving adenosine triphosphate-binding cassette (ABC) and solute carrier transporters are altered decreasing chemosensitivity of the cells. Human proteins of ABC family are divided into seven subfamilies (A to G) depending on the domain organization. Three protein members are well known for their MDR, namely P-glycoprotein (P-gp, MDR1, a phosphorylated and glycosylated 170 kDa protein of 1280 amino acids), multidrug resistance associated protein 1 (MRP1), and breast cancer resistance protein (BCRP) effluxing vinca alkaloids, taxanes, podophyllotoxins and anthracyclines group of drugs [4]. Normal expression of MDR1 becomes extensively high when colon, liver and kidney cells are cancerous reducing drug transport inside the cells. Lung, prostate and breast cells overexpress MRP1 or BRCP protecting cells from cell death. BRCP inhibitory hormonal therapy with 17 β estradiol and Gefitinib have shown better sensitivity to chemotherapy following combining these drug with anticancer drugs. P-glycoprotein alterations by kinase linked signaling molecules are also studied. Estrogen down-regulates P-gp expressions in Estrogen Receptor Positive breast carcinoma, conversely MAPK proteins like c-Raf, MEK1/2, ERK1/2 and growth factors such as EGF and FGF up-regulate its expression [4,5].
2. Altered target proteins and enzyme pathway
Topoisomerases regulating the topographical state during DNA replication and transcription is a target for anthracyclines. Relationship between mRNA expression of topoisomerase II and response to anthracylines demonstrates that increased expression of the enzyme leads to increased sensitivity to the drug. In some breast cancer cases amplification of genes showing the effects of reduced sensitivity have been demonstrated [6].
3. Reduction in activation of drug
Complex enzymatic proteins can partially degrade, modify, or complex the drug with proteins to initiate metabolic activation of drug for clinical efficacy. These systems can develop resistance to reduce therapeutic efficacy in vivo. In case of cytarabine, which is activated via multiple phosphorylation processes to convert into AraC-triphosphate, the conversion may be disrupted owning to mutation in pathway. Other examples include CYP 450, glutathione-S-transferases (GST) family that protects the cells from attacks of electrophiles [5].
4. Altered expression of enzymes and enhancement of drug degradation in vivo
Increase in the breakdown of drug and elimination by kidneys are important consideration in case of alteration in cancer chemotherapy. CYP family comprising Class I and Class II enzymes take active roles in anticancer drug disposition in vivo. Class II compounds are responsible for drug metabolism and hence are major targets for altered mutation while Class I enzymes participate in pro carcinogen metabolism and alteration of capabilities produce resistance in vivo. GSTs protect the cells from electrophile attack to enhance survival and may assist in resistance through direct detoxification and inhibition of mitogen-activated protein kinase pathway. The inappropriate expression of aldehyde dehydrogenases that participate in intracellular detoxification of cyclophosphamide and conversion of aldophosphamide into inactive forms may be a potential resistance factor [6,7].
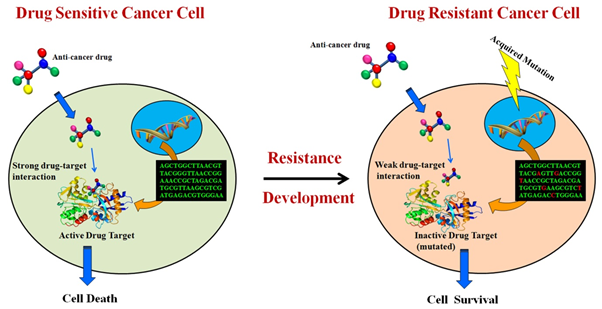
Fig 3: Development of drug resistance within cancer cells (Image source: http://crdd.osdd.net/raghava/cancerdr/)
5. Drug inactivation after conjugation with cell metabolites
Platinum based anticancer drugs such as cisplatin, oxaloplatin and carboplatin are taken up by the cells and Pt atom forms Pt-DNA adducts binding covalently in N-7 position of Guanine. It forms 1,2 or 1,3 intrastand cross links and interstand cross links allowing cell death to occur. The absorption of platinum is mediated by a copper transporter, therefore intracellular copper binding thiol containing proteins like GSH and metallothioneins bind not only with copper but also with platinum producing Pt(GS)2 decreasing its activity.
6. Drug interaction and cross resistance
An important feature of drug resistance is that resistance over a single drug may initiate resistance to other drugs. For instance, loss of drug transporters due to alteration of proteins by one drug may cause reduced influx of drug molecules inside the cells, thereby reducing responsiveness. Elevation of ABC transporters may influence the efficacy of many other drugs leading to poor chemotherapy response [7].
7. Enhanced DNA and nucleotide excision repair
Adduct formation causing lesion to the nucleotide sequence often leads to degradation of DNA in vivo as a mean of chemotherapy response. Pt-DNA adducts formation by platinum based anti cancer compounds are hindered by nucleotide excision repair and homologous recombination, reversing the therapeutic efficacy of platinum based compounds. The DNA repair by O6-methylguanine DNA methyltransferases (MGMT) is induced by many drugs. MGMT repairs alkylated nucleotides before mismatch identification and protect stem cells from alkylating agents. To target specifically the MGMT present in cancer cells without affecting normal cells, MGMT promoter CpG biomarker is targeted in therapy but in this case limitations such as uncertain prognosis and genetic instability cannot be avoided. In breast and ovarian cancers mutations in BRCA1 and BRCA2 genes, meditates DNA double-strand break repair. Additionally poly (ADP-ribose) polymerase protein which is responsible for such repair can be targeted with PARP inhibitors such as iniparib and veliparib. Impairment of DNA damage response (DDR) genes and epigenetic silencing are mechanism underlying combination therapy to make cancer cells responsive towards therapy [5,8].
8. Apoptosis defects due to molecular interactions
Apotosis and autophagy are two compensatory and antagonistic regulatory events that play an enormous role in cell mediated damage following chemotherapy. The two established pathways of apoptosis include intrinsic pathway mediated by mitochondria involving B-cell lymphoma (Bcl-2), caspase family and Akt and extrinsic pathway involving apoptotic surface receptors on cells. Resistance could by triggered by inactivation and mutation in genes coding apoptotic proteins such as p53, or enhancement of anti apoptotic proteins such as Bcl-2 limiting therapeutic efficacy of doxorubicin in advanced breast carcinoma and anthracyclines in mouse sarcoma tumor model. Recombinant forms of tumor necrosis factors related to apoptosis inducing ligand (TRAIL) along with cytotoxic drugs have shown to produce caspase 8 mediated cytotoxicity enhancing chemosensitivity [9].
ORIGIN OF RESISTANT CELLS
Alteration of both genetic and epigenetic modification over time can develop the cancer cells in a multistage process. As a result enhancement of drug resistant cancer cells in heterogeneous cell populations can modulate the resistance in cancer progenitor and adult cancer cells.
1. Cell heterogeneity
A single tumor can comprise heterogeneous populations of cells with distinct genetic fingerprints known as polygenomic subpopulations, as revealed in a study of epithelial breast cancer showing 97% of intra tumor diverseness. Cancer cell selection obeys Darwinian law of evolution, and under therapeutic pressure the adaptive or resistant cells will be selected for clonal selection populating tumor comprising of highly resistant cells. Mutated drug binding site can be seen in cells as well as acquisition of stochastic alteration within cells provides survival advantages are the two possible mechanisms of resistance. In addition, survival of these cells contribute to pathology over time by formation of tumors in distinct organs [5,7].
2. Cancer Stem Cell (CSC) model
In this model, it is demonstrated that rare populations of cancer stem cells possess properties to initiate a new tumor. CSCs are diverged from normal stem cells through deregulation of self-renewal pathways, and act as causative agents of altered drug transport and enzymatic activities, over-expression of cell cycle proteins, initiating resistance in a quiescent nature making them difficult to target with drug [7].
3. Environment Mediated Drug Resistance (EMDR) model
In this model resistance emerges as the cancer cells enter into a dormant stage to circumvent the effects of given therapy. Under drug pressure, they undergo constant alteration in their genetic makeup ultimately achieving a permanent acquired resistance phenotype altering their surrounding microenvironments. Soluble factors such as chemokines, cytokines or growth factors secreted by fibroblast can induce gene transcription. One study reveals the usage of IL-6 in this regard in both in vitro and in vivo models. Cell adhesion mediated drug resistance occurs with adhesion of integrins to stromal fibroblasts surrounding extracellular matrix. Furthermore it can be a effective target for combination therapy comprising cytotoxic drugs along with inhibitors of EMDR pathways. In another study significantly reduced myeloma growth were observed in combination therapy of melphalan and α-4 antibody of integrin (natalizumab) [7,9].
CONCLUSION
Clinical usage of conventional chemotherapeutic agents remains in an area of regular studies based on data generation from different parts of world. Cancer drug resistance is a complex phenomena involving drug inactivation, altered membrane transport and enzymatic activities, DNA damage repair and apoptosis inhibition. In order to apply a justified combination therapy, these targets are to be considered for designing inhibitor to specific gene. Along with si RNA and mi RNA based therapeutics; various antibodies have shown their proficiency in gene silencing and thereby increased chemosensitivity of the cells. Therefore it is important to continue efforts to demonstrate the mechanisms of anticancer drug resistance and to identify novel therapeutics in future days.
References
1. Globocan 2012, http://globocan.iarc.fr/Default.aspx Accessed on 18th December, 2016
2. "Over 17 lakh new cancer cases in India by 2020: ICMR" http;//www.mid-day.com/articles/over-17-lakh-new-cancer-cases-in-india-by-2020-icmr/17248152 Accessed on 20th December, 2016.
3. Chorawala MR, Oza PM, Shah GB (March 2012). "Mechanisms of anticancer drugs resistance: an overview". Int. J Pharm. Sci Drug Res 4(1): 01-9.
4. Liu FS (September 2009). "Mechanisms of chemotherapeutic drug resistance in cancer therapy-a quick review". Taiwan J Obstet Gynecol 48(3): 239-44.
5. Housman G, Byler S, Heerboth S et al. (September 2014). "Drug resistances in cancer: an overview". Cancers 6(1): 1769-92.
6. Ringborg U, Platz A (July 2009). "Chemotherapy resistance mechanisms". Acta Oncol. 35(5): 76-80.
7. Zahreddine H, Borden LB (March 2013). "Mechanisms and insights into drug resistance in cancer". Frontiers Pharmacol 4(28): 1-8.
8. Friedman R (February, 2016). "Drug resistance in cancer: molecular evolution and compensatory proliferation". Oncotarget 7(11): 11746-55.
About Author / Additional Info:
I am currently pursuing M.Pharm in Pharmaceutics from Jadavpur University.